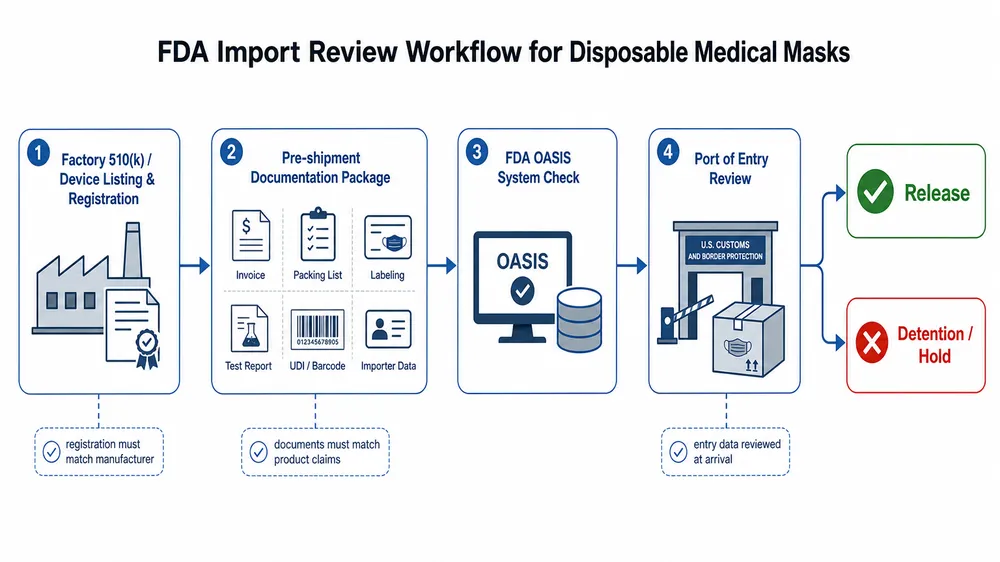
Most FDA import failures for disposable medical masks don't happen because the product is bad. They happen because the paperwork doesn't match the product, the product doesn't match the claimed standard, or the factory behind it was never registered to begin with.
We've been manufacturing and exporting disposable medical masks since 2012. We've seen what happens when a shipment gets detained at a US port of entry — and we've spent years building the documentation infrastructure that prevents it. This guide covers what FDA import review actually checks, what documents your supplier needs to hand you before the order ships, and how to read a mask spec sheet so you're not guessing at BFE numbers.

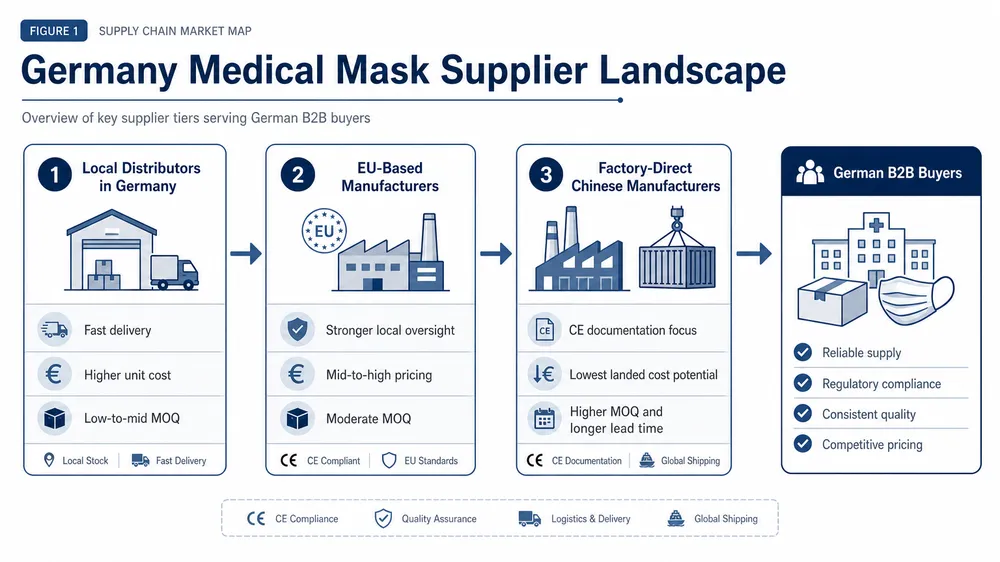
What FDA import review actually checks
The FDA treats disposable medical masks as Class II medical devices. That classification matters because it means your supplier needs a 510(k) clearance — not just a CE mark, not just an ISO certificate, not just a test report from a third-party lab.
When a shipment arrives at a US port of entry, FDA's OASIS system flags it for review. The system checks whether the manufacturer is registered with FDA and whether the device is listed. If the factory isn't in the FDA establishment registration database, the shipment can be detained on arrival — before anyone opens a box.
What triggers a detention or refusal:
- No FDA establishment registration — the factory has no record in the FDA database
- No 510(k) clearance number — the product is listed as a medical device but has no cleared predicate
- Labeling non-compliance — missing intended use statement, missing manufacturer name and address, or claims that exceed the cleared indication
- Documentation mismatch — the test reports reference a different product configuration than what's in the shipment
- Meltblown fabric substitution — the BFE/PFE values on the CoC don't match the actual filtration performance of the batch shipped
That last point is where most buyers get caught off guard. A supplier can show you a BFE ≥95% test report from six months ago. If they switched meltblown fabric suppliers since then — which happens more than buyers realize — the current batch may not hit that spec. We'll come back to this.
The documentation package a compliant shipment needs
Before any order ships, you should have these documents in hand. Not on request after arrival. Before the container leaves the factory.
| Document | What it confirms | Where to verify |
|---|---|---|
| FDA 510(k) clearance number | Product is cleared as a Class II medical device | FDA 510(k) database (accessdata.fda.gov) |
| FDA establishment registration number | Factory is registered with FDA | FDA FEI database |
| ASTM F2100 test report | BFE, PFE, Delta-P, fluid resistance, flammability | Accredited lab (Nelson Labs, SGS, Intertek) |
| Certificate of Conformance (CoC) | Batch-level confirmation of spec compliance | Issued by factory QC |
| Packing list with lot numbers | Traceability from shipment to production batch | Factory documentation |
| Labeling proof | Confirms label content matches FDA cleared indication | Factory or brand owner |
| ISO 13485 certificate | Quality management system for medical devices | Issuing certification body |
(The CoC is often treated as a formality. It isn't. If the CoC lot number doesn't trace back to a production batch with corresponding in-process QC records, it's a piece of paper, not evidence.)
A supplier who hesitates on any of these documents before you place an order is telling you something. We send this package as standard with every order confirmation — it's not a special request.

How to read a mask spec sheet: BFE, PFE, Delta-P, and ASTM F2100 levels
ASTM F2100 defines three performance levels for medical face masks. The level determines which application the mask is appropriate for — and which markets will accept it.
| ASTM F2100 Level | BFE | PFE | Delta-P (Pa/cm²) | Fluid Resistance | Typical application |
|---|---|---|---|---|---|
| Level 1 | ≥95% | ≥95% | <4.0 | 80 mmHg | Low-risk clinical, general medical |
| Level 2 | ≥98% | ≥98% | <5.0 | 120 mmHg | Moderate-risk surgical, aerosol exposure |
| Level 3 | ≥98% | ≥98% | <5.0 | 160 mmHg | High-risk surgical, high fluid exposure |
BFE (Bacterial Filtration Efficiency) and PFE (Particulate Filtration Efficiency) are the numbers most buyers focus on. Delta-P is the one most buyers ignore — and it matters for compliance.
Delta-P measures airflow resistance through the mask. A mask with very high BFE but Delta-P above the ASTM threshold fails the standard regardless of filtration performance. We see this with masks that use overly dense meltblown layers to hit filtration numbers without balancing breathability. The mask passes BFE on paper but fails the full ASTM F2100 panel.
My signature quote on this: BFE and PFE numbers only mean something if you know what went into the meltblown layer. A 95% BFE result from a well-controlled meltblown batch is reliable. The same number from a factory that buys meltblown on the spot market from whoever is cheapest that week is not.
Fluid resistance is the third variable that separates Level 1 from Level 3. If your buyers are supplying surgical environments with high fluid exposure, Level 1 won't pass their procurement audit. Confirm the fluid resistance rating before you commit to a product.

Why in-house meltblown production changes your BFE risk profile
This is the part of disposable medical mask sourcing that most guides skip entirely.
The meltblown nonwoven layer is the filtration core of a disposable medical mask. It's what determines BFE and PFE performance. Most factories don't make their own meltblown — they buy it from external suppliers. That's not inherently a problem, but it creates a supply chain variable that directly affects spec consistency across your orders.
When a factory sources meltblown externally, their BFE performance is only as stable as their fabric supplier's production consistency. If the fabric supplier changes their polymer blend, adjusts their extrusion parameters, or substitutes a different grade during a shortage, the filtration performance of the finished mask changes — even if the factory's own process stays identical. The factory may not even know until a batch fails QC.
We produce our own meltblown nonwoven fabric in-house. That means we control the fiber diameter, the basis weight, the electrostatic charge treatment, and the lot-to-lot consistency of the filtration layer. When we issue a CoC showing BFE ≥98%, that number is backed by in-process meltblown batch testing, not by a certificate from a third-party fabric roll we received last week.
For buyers placing repeat orders — distributors building a private-label program, procurement teams running annual supply contracts — this matters more than the initial test report. The first shipment's BFE is easy to verify. The tenth shipment's BFE is where in-house meltblown control shows its value.
(We switched to full in-house meltblown production in 2019. Before that, we dual-sourced. The batch-to-batch BFE variance we saw from external suppliers was the reason we made the investment.)
Factory qualification: what to check before you place an order
A test report from an accredited lab tells you what one batch of masks performed. It doesn't tell you whether the factory can reproduce that performance at scale, across multiple production runs, under the conditions of your actual order.
These are the factory-level factors that determine whether your spec holds across a full program:
Cleanroom manufacturing environment. Disposable medical masks produced for hospital and institutional procurement need to be manufactured in a controlled environment. ISO 13485 requires documented environmental controls. Our facility runs a Class 100,000 (ISO 8) cleanroom — that's the standard that satisfies hospital procurement facility requirements in most markets. Ask your supplier for their cleanroom classification and the monitoring records.
ISO 13485 certification. ISO 9001 is a general quality management standard. ISO 13485 is specific to medical devices and includes requirements for sterile product controls, traceability, and post-market surveillance. For FDA-registered medical masks, ISO 13485 is the baseline expectation. A factory with only ISO 9001 is not operating under a medical device QMS.
In-house QC lab with ASTM-capable testing equipment. A factory that can only show you third-party test reports — and can't run BFE, PFE, or Delta-P in-house — has no way to catch a filtration problem before it ships. We run BFE, PFE, and Delta-P testing in our own lab on every production batch. That's not a marketing claim; it's the only way to catch a meltblown variance before it becomes your problem at the port.
Production line count and capacity. 6 fully automated production lines at 120 million pieces annual capacity means your order doesn't queue behind a larger customer's run. For buyers placing 50,000–500,000 piece orders, this is a lead time question, not just a scale question.
Audit readiness. Ask whether the factory has passed a third-party audit in the last 12 months. We hold SGS audit certification alongside our ISO 13485 and ISO 9001:2015 registrations. An audited factory has documented its processes in a way that survives external scrutiny — which is exactly what FDA import review and hospital procurement audits require.
EU import parallel: CE MDR and what changed post-2021
If you're importing into the EU as well as the US, the regulatory picture changed significantly in May 2021 when the EU Medical Device Regulation (EU MDR 2017/745) replaced the older MDD framework.
Under EU MDR, medical face masks classified as medical devices require CE marking under the new regulation — not the legacy MDD CE mark. The key differences that affect importers:
- Notified Body involvement is now required for Class I medical devices with a measuring function or sterile presentation, and for Class IIa and above. Disposable medical masks typically fall under Class I, but the classification depends on the intended use claim on the label.
- Technical documentation requirements are more extensive under MDR. The manufacturer must maintain a complete technical file including clinical evaluation, risk management documentation, and post-market surveillance plan.
- Importer obligations under EU MDR are more explicit. As the EU importer of record, you are responsible for verifying that the manufacturer has fulfilled their obligations — including CE marking, technical documentation, and registration in the EUDAMED database.
We hold CE marking under EU MDR for our disposable medical mask range. For buyers importing into the EU, we can provide the technical documentation summary and EUDAMED registration reference on request.
(One thing that catches EU importers off guard: a CE mark issued under the old MDD before May 2021 is no longer valid for new market placements. If your current supplier's CE certificate predates the MDR transition and hasn't been renewed, you're importing on an expired certification.)
How to structure a trial order before committing to volume
The right way to validate a new disposable medical mask supplier is not to place a 500,000-piece order and hope the spec holds. It's to structure a trial order that gives you enough product to test, enough documentation to verify, and enough production data to assess consistency.
A workable trial order structure:
- Order 50,000–100,000 pieces — enough to run your own independent BFE/PFE testing on a statistically meaningful sample, and enough to evaluate packaging, labeling, and dimensional consistency.
- Request lot-specific test reports — not the factory's standard product test report, but a report tied to the specific production lot in your trial order. This confirms the factory is testing each batch, not relying on a historical result.
- Verify the 510(k) number independently — search the FDA 510(k) database before the order ships. The clearance number should be searchable and should match the product description in your order.
- Check the meltblown source — ask directly whether the factory produces meltblown in-house or sources externally. If external, ask for the fabric supplier's name and whether they have a qualified supplier approval process.
- Review the labeling against FDA cleared indication — the label on the mask must match the intended use statement in the 510(k) clearance. Any deviation is a labeling non-compliance that can trigger detention.
Our standard MOQ for Disposable Medical Masks is 50,000 pieces for standard SKUs — sized specifically to support trial orders before buyers commit to a full program. Most of our North American and European buyers start with a trial run, run their own lab verification, and then move to quarterly supply contracts.
For buyers evaluating 3-ply disposable medical masks or 4-ply disposable medical masks, the trial order process is the same — the 4-ply configuration adds a second meltblown layer, which affects both BFE performance and Delta-P, so independent testing on the specific configuration you're ordering is worth the cost.
Frequently asked questions
Does a CE mark substitute for FDA 510(k) clearance for US imports?
No. CE marking under EU MDR and FDA 510(k) clearance are separate regulatory pathways with no mutual recognition. A CE-marked mask can be detained at a US port of entry if the manufacturer lacks FDA establishment registration and 510(k) clearance. The two certifications are both required if you're selling into both markets — they don't substitute for each other.
What's the difference between a surgical mask and a medical mask under FDA classification?
FDA classifies surgical masks (product code FPA) and medical face masks (product code MSH) as separate device types with different 510(k) predicates. The distinction matters for labeling: a mask labeled "surgical mask" must meet the fluid resistance requirements associated with that classification. If your supplier's 510(k) clearance is for a medical face mask but the product is labeled as a surgical mask, that's a labeling non-compliance. Confirm the product code in the 510(k) clearance matches the label claim.
How often should ASTM F2100 test reports be renewed?
FDA doesn't mandate a fixed renewal interval for 510(k)-cleared devices, but most institutional procurement standards and hospital GPO contracts require test reports dated within 12–24 months. More practically: if your supplier has changed their meltblown source, production line configuration, or mask construction since the last test report, that report no longer reflects the current product. Request a new report tied to the current production configuration, not the original 510(k) submission.
What MOQ is realistic for a compliance-validated trial order?
50,000 pieces is the practical floor for a meaningful trial. Below that, you don't have enough product to run independent lab testing on a statistically valid sample and still have inventory to sell. At 50,000 pieces, you can send 200–300 units to an accredited lab (Nelson Labs or SGS are the standard choices for ASTM F2100), keep the remainder for market testing, and have enough lot-specific documentation to evaluate the factory's QC process.
Can a factory's ISO 13485 certificate be verified independently?
Yes. ISO 13485 certificates are issued by accredited certification bodies (BSI, TÜV, SGS, Bureau Veritas, and others). The issuing body maintains a public or request-accessible registry of current certificates. Ask your supplier for the certificate number and the issuing body's name, then verify directly with that body. An expired or suspended certificate won't appear as current in the registry.
What does "Class 100,000 cleanroom" mean for mask quality?
Class 100,000 (ISO 8) means the manufacturing environment is controlled to no more than 100,000 particles ≥0.5 microns per cubic foot of air. For disposable medical masks, this matters for two reasons: it reduces the risk of particulate contamination in the finished product, and it's the facility standard that hospital and institutional procurement audits typically require. A factory producing medical masks in an uncontrolled environment can't credibly claim the product meets hospital-grade procurement specifications, regardless of what the test report says.
If you're evaluating suppliers for a US or EU import program and want to see eztio's full documentation package — 510(k) registration, ASTM F2100 test reports, ISO 13485 certificate, and CE MDR documentation — send us your RFQ with your target market, required filtration grade, and order volume. We'll respond with a compliant product recommendation and the complete documentation set.